



RT-qPCR är utvecklad från vanlig PCR-teknik.Den lägger till fluorescerande kemikalier (fluorescerande färgämnen eller fluorescerande prober) till det traditionella PCR-reaktionssystemet och detekterar PCR-glödgnings- och förlängningsprocessen i realtid enligt deras olika luminiscerande mekanismer.Fluorescerande signalförändringar i mediet används för att beräkna mängden produktförändring i varje PCR-cykel.För närvarande är de vanligaste metoderna fluorescerande färgmetod och sondmetod.
Fluorescerande färgningsmetod:
Vissa fluorescerande färgämnen, såsom SYBR Green Ⅰ, PicoGreen, BEBO, etc., avger inte ljus av sig själva, men avger fluorescens efter att de har bindits till det mindre spåret i dsDNA.Därför, i början av PCR-reaktionen, kan maskinen inte detektera den fluorescerande signalen.När reaktionen fortsätter till hybridiseringsförlängningen (tvåstegsmetoden) eller förlängningssteget (trestegsmetoden), öppnas de dubbla strängarna vid denna tidpunkt, och det nya DNA-polymeraset Under strängsyntes kombineras fluorescerande molekyler i dsDNA-minor groove och avger fluorescens.När antalet PCR-cykler ökar, kombineras fler och fler färgämnen med dsDNA, och den fluorescerande signalen förstärks också kontinuerligt.Ta SYBR Green Ⅰ som ett exempel.
Sondmetod:
Taqman-sonden är den mest använda hydrolyssonden.Det finns en fluorescerande grupp vid 5′-änden av sonden, vanligtvis FAM.Själva sonden är en sekvens som är komplementär till målgenen.Det finns en fluorescerande släckningsgrupp vid 3′-änden av fluoroforen.Enligt principen för fluorescensresonansenergiöverföring (Förster resonance energy transfer, FRET), när reporterfluorescensgruppen (donatorfluorescerande molekylen) och den släckande fluorescerande gruppen (acceptorfluorescerande molekylen) När excitationsspektrumet överlappar och avståndet är mycket nära (7-10 fluorescerande molekylen) acceptormolekylen, medan autofluorescensen försvagas.Därför, i början av PCR-reaktionen, när sonden är fri och intakt i systemet, kommer den fluorescerande reportergruppen inte att avge fluorescens.Vid hybridisering binder primern och sonden till mallen.Under förlängningssteget syntetiserar polymeraset kontinuerligt nya kedjor.DNA-polymeras har 5′-3′ exonukleasaktivitet.När det når sonden kommer DNA-polymeraset att hydrolysera sonden från mallen, separera reporterfluorescerande gruppen från quencher-fluorescerande gruppen och frigöra den fluorescerande signalen.Eftersom det finns ett en-till-en-förhållande mellan sonden och mallen, är sondmetoden överlägsen färgmetoden när det gäller testets noggrannhet och känslighet.
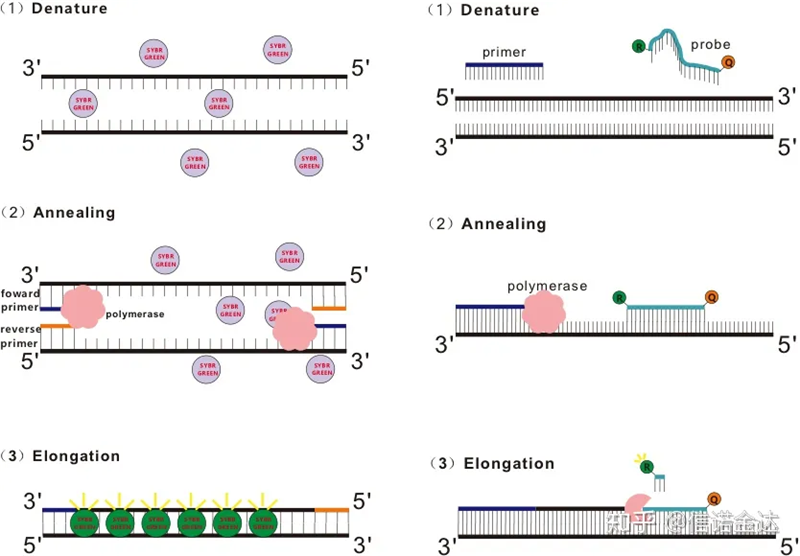
Fig 1 Principen för QRT-PCR
Primer design
Principer:
Primerna bör utformas i den konserverade regionen av nukleinsyraserien och ha specificitet.
Det är bäst att använda cDNA-sekvens, och mRNA-sekvens är också acceptabel.Om inte, ta reda på cd-regionens design av DNA-sekvensen.
Längden på den fluorescerande kvantitativa produkten är 80-150 bp, den längsta är 300 bp, primerlängden är i allmänhet mellan 17-25 baser, och skillnaden mellan uppströms- och nedströmsprimrarna bör inte vara för stor.
G+C-halten är mellan 40% och 60% och 45-55% är bäst.
TM-värdet är mellan 58-62 grader.
Försök att undvika primerdimerer och självdimerer, (visas inte fler än 4 par på varandra följande komplementära baser) hårnålsstruktur, om det är oundvikligt, gör ΔG<4,5kJ/mol* Om du inte kan säkerställa att gDNA har tagits bort under omvänd transkription Rengör, är det bäst att designa primrarna för intronen och G′-änden, kan inte vara modifierad, G′-regionen *3 kan inte modifieras. /C, A/G kontinuerlig struktur (2-3) primers och icke-
specifik Homologin för den heterogent amplifierade sekvensen är företrädesvis mindre än 70 % eller har 8 komplementär bashomologi.
Databas:
CottonFGD sök med nyckelord
Primer design:
IDT-qPCR primer design
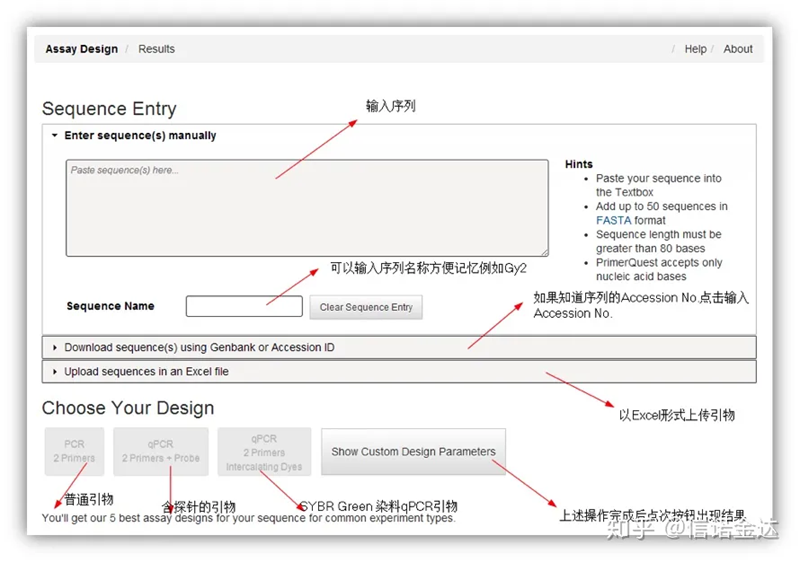
Fig2 IDT online primer design verktygssida
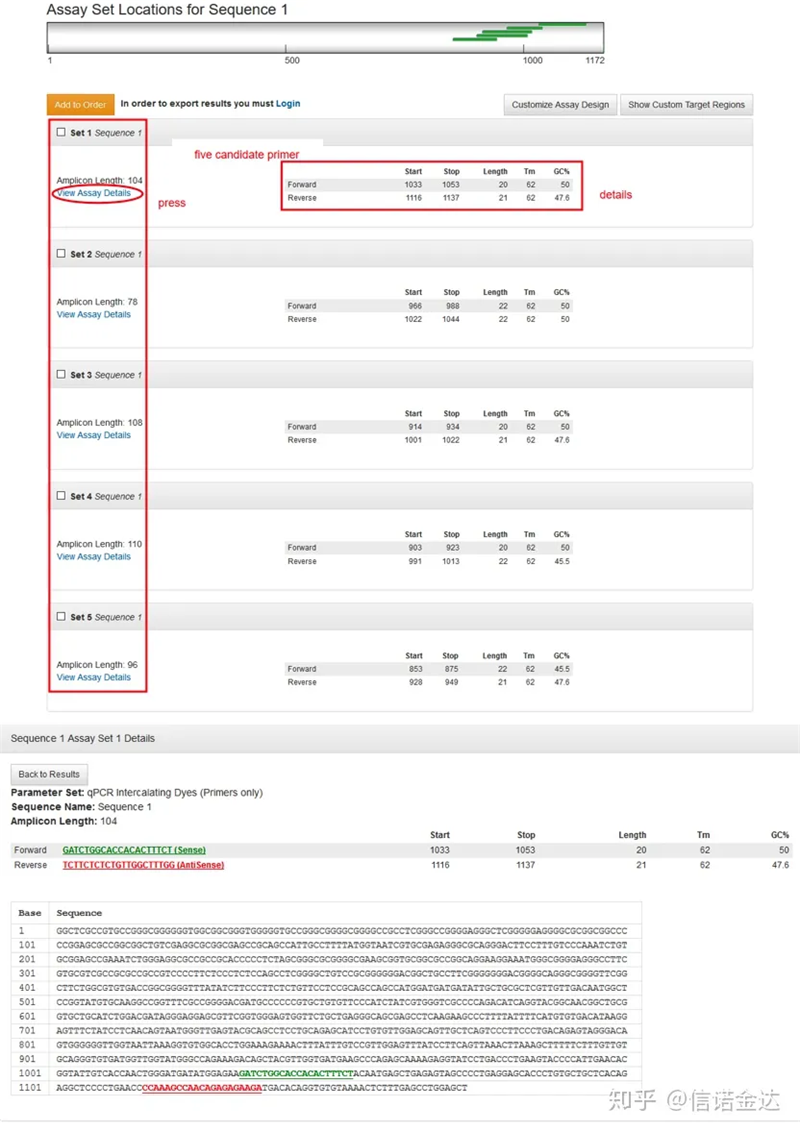
Bild 3 resultatsida
Design av lncRNA-primrar:
lncRNA:samma steg som mRNA.
miRNA:Principen för stam-loop-metoden: Eftersom alla miRNA är korta sekvenser på cirka 23 nt, kan direkt PCR-detektion inte utföras, så stam-loop-sekvensverktyget används.Stam-loop-sekvensen är ett enkelsträngat DNA på cirka 50 nt, som kan bilda en hårnålsstruktur av sig själv.3 'Sluten kan utformas som en sekvens som är komplementär till miRNA-partiella fragmentet, sedan kan mål-miRNA:t kopplas till stam-loop-sekvensen under omvänd transkription, och den totala längden kan nå 70 bp, vilket är i linje med längden på den amplifierade produkten bestäms av qPCR.Tailing miRNA primer design.
Amplifieringsspecifik detektion:
Online sprängdatabas: CottonFGD sprängning efter sekvenslikhet
Lokal blast: Se att använda Blast+ för att göra lokal blast, linux och macos kan direkt upprätta en lokal databas, win10-system kan också göras efter installation av ubuntu bash.Skapa lokal blast-databas och lokal blast;öppna ubuntu bash på win10.
Observera: Bomull i högland och bomull på havsöar är tetraploida grödor, så resultatet av sprängningen blir ofta två eller flera matchningar.Tidigare har man troligen hittat två homologa gener med endast ett fåtal SNP-skillnader att använda NAU-cd-skivor som en databas för att utföra blast.Vanligtvis kan de två homologa generna inte separeras genom primerdesign, så de behandlas som samma.Om det finns en uppenbar indel, är primern vanligtvis utformad på indel, men detta kan leda till den sekundära strukturen av primern. Den fria energin blir högre, vilket leder till en minskning av amplifieringseffektiviteten, men detta är oundvikligt.
Detektion av primer sekundär struktur:
Steg:öppna oligo 7 → mata in mallsekvens → stäng underfönster → spara → lokalisera primer på mall, tryck på ctrl+D för att ställa in primerlängd → analysera olika sekundära strukturer, såsom självdimeriseringskropp, heterodimer, hårnål, missmatch etc. De två sista bilderna i figur 4 är testresultaten av primrarna.Resultatet av den främre primern är bra, det finns ingen uppenbar dimer- och hårnålsstruktur, inga kontinuerliga komplementära baser, och det absoluta värdet av fri energi är mindre än 4,5, medan den bakre primern visar kontinuerlig. De 6 baserna är komplementära och den fria energin är 8,8;dessutom uppträder en allvarligare dimer vid 3'-änden och en dimer med 4 på varandra följande baser.Även om den fria energin inte är hög kan 3'-dimer Chl allvarligt påverka amplifieringsspecificiteten och amplifieringseffektiviteten.Dessutom är det nödvändigt att kontrollera hårnålar, heterodimerer och felmatchningar.
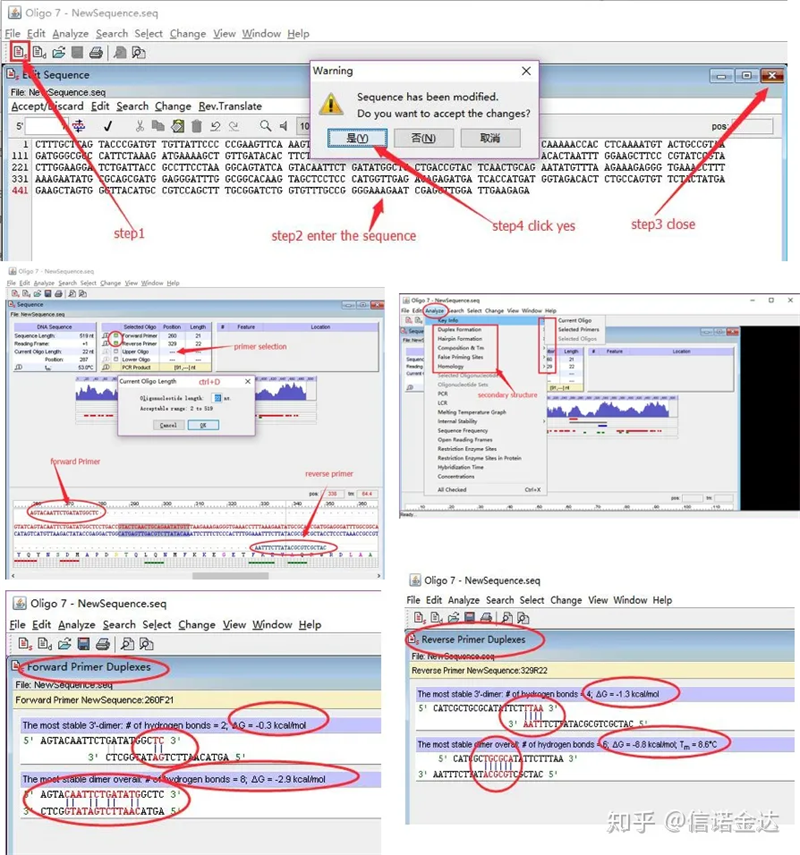
Fig3 oligo7 detektionsresultat
Detektering av amplifieringseffektivitet:
Amplifieringseffektiviteten för PCR-reaktionen påverkar allvarligt PCR-resultaten.Även i qRT-PCR är amplifieringseffektiviteten särskilt viktig för de kvantitativa resultaten.Ta bort andra ämnen, maskiner och protokoll i reaktionsbufferten.Kvaliteten på primrarna har också ett stort inflytande på amplifieringseffektiviteten för qRT-PCR.För att säkerställa noggrannheten av resultaten måste både den relativa fluorescenskvantifieringen och den absoluta fluorescenskvantifieringen detektera amplifieringseffektiviteten för primrarna.Det är känt att den effektiva qRT-PCR-amplifieringseffektiviteten är mellan 85 % och 115 %.Det finns två metoder:
1. Standardkurvmetod:
a.Blanda cDNA
b.Gradientutspädning
c.qPCR
d.Linjär regressionsekvation för att beräkna amplifieringseffektiviteten
2. LinRegPCR
LinRegPCR är ett program för analys av RT-PCR-data i realtid, även kallat kvantitativ PCR-data (qPCR) baserad på SYBR Green eller liknande kemi.Programmet använder icke-baslinjekorrigerade data, utför en baslinjekorrigering på varje prov separat, bestämmer ett linjäritetsfönster och använder sedan linjär regressionsanalys för att passa en rak linje genom PCR-datauppsättningen.Från lutningen av denna linje beräknas PCR-effektiviteten för varje enskilt prov.Den genomsnittliga PCR-effektiviteten per amplikon och Ct-värdet per prov används för att beräkna en startkoncentration per prov, uttryckt i godtyckliga fluorescensenheter.Datainmatning och -utdata sker via ett Excel-kalkylblad.Endast prov
blandning krävs, ingen gradient
steg krävs:(Ta Bole CFX96 som ett exempel, inte riktigt Maskin med tydlig ABI)
experimentera:det är ett standard qPCR-experiment.
qPCR-datautgång:LinRegPCR kan känna igen två former av utdatafiler: RDML eller kvantifieringsförstärkningsresultat.I själva verket är det realtidsdetekteringsvärdet för cykelnumret och fluorescenssignalen av maskinen, och amplifieringen erhålls genom att analysera fluorescensändringsvärdet för det linjära segmentets effektivitet.
Dataurval: I teorin bör RDML-värdet vara användbart.Det uppskattas att problemet med min dator är att programvaran inte kan känna igen RDML, så jag har excel-utgångsvärdet som originaldata.Det rekommenderas att utföra en grov screening av data först, såsom misslyckande med att lägga till prover, etc. Punkterna kan raderas i utdata (naturligtvis kan du inte radera dem, LinRegPCR kommer att ignorera dessa punkter i ett senare skede)
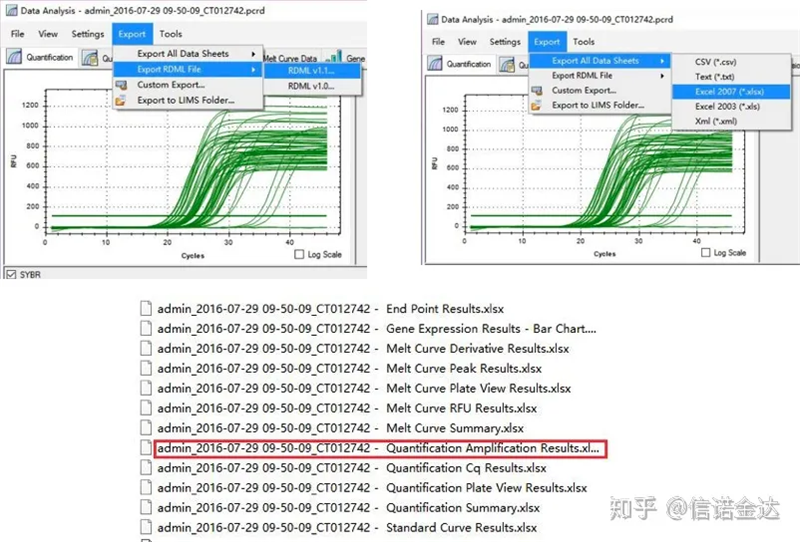
Fig5 qPCR-dataexport
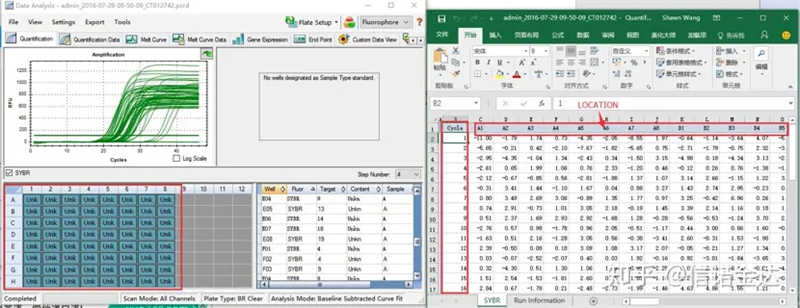
Fig 6 urval av kandidatprover
Dataingång:Öppna kvalificeringsförstärkningsresultat.xls, → öppna LinRegPCR → fil → läs från excel → välj parametrar som visas i figur 7 → OK → klicka på bestämma baslinjer
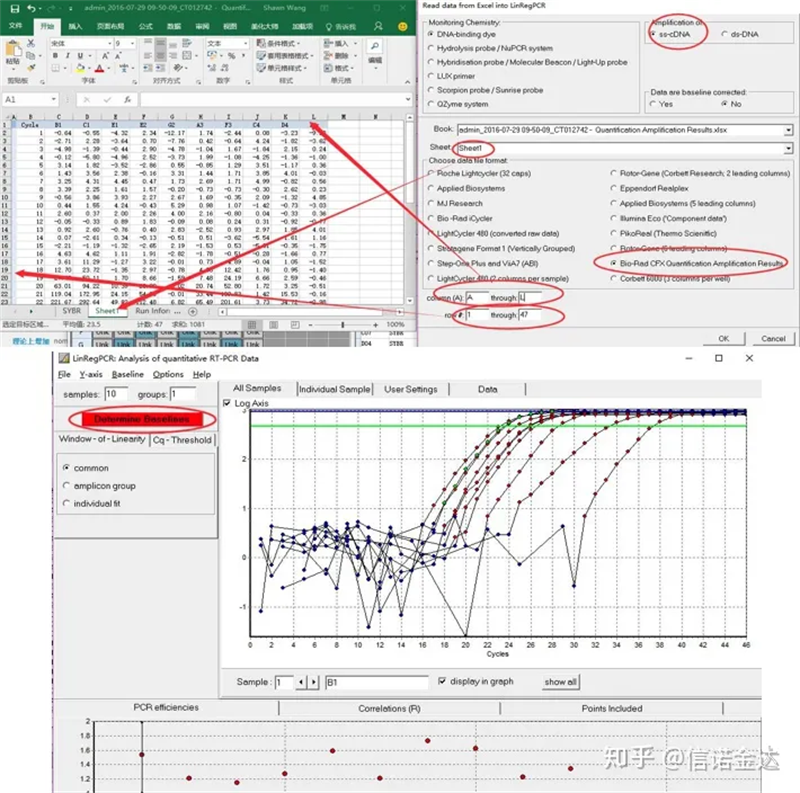
Fig7 steg för linRegPCR-datainmatning
Resultat:Om det inte finns någon upprepning krävs ingen gruppering.Om det förekommer upprepning kan grupperingen redigeras i provgrupperingen, och namnet på genen skrivs in i identifieraren, och sedan grupperas samma gen automatiskt.Klicka slutligen på filen, exportera excel och se resultaten.Amplifieringseffektiviteten och R2-resultaten för varje brunn kommer att visas.För det andra, om du delar upp i grupper, kommer den korrigerade genomsnittliga förstärkningseffektiviteten att visas.Se till att amplifieringseffektiviteten för varje primer är mellan 85 % och 115 %.Om den är för stor eller för liten betyder det att primerns amplifieringseffektivitet är dålig.
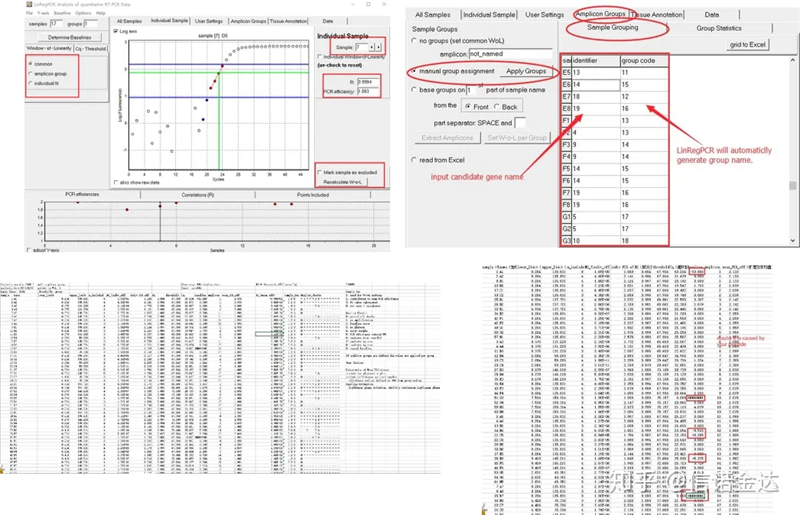
Fig 8 Resultat och datautgång
Experimentell process:
RNA kvalitetskrav:
Renhet:1.72,0 indikerar att det kan finnas kvarvarande isotiocyanat.Ren nukleinsyra A260/A230 bör vara cirka 2. Om det finns en stark absorption vid 230 nm, indikerar det att det finns organiska föreningar såsom fenatjoner.Dessutom kan det detekteras genom 1,5 % agarosgelelektrofores.Peka på markören, eftersom ssRNA inte har någon denaturering och molekylviktslogaritmen inte har ett linjärt samband, och molekylvikten kan inte uttryckas korrekt.Koncentration: Teoretisktintemindre än 100 ng/ul, om koncentrationen är för låg är renheten i allmänhet låg, inte hög
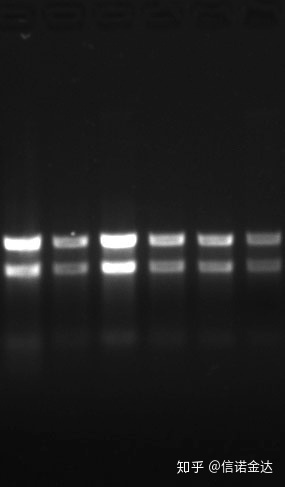
Fig 9 RNA-gel
Dessutom, om provet är värdefullt och RNA-koncentrationen är hög, rekommenderas det att alikvotera det efter extraktion och späda RNA:t till en slutlig koncentration av 100-300 ng/ul för omvänd transkription.Iprocessen med omvänd transkription, när mRNA transkriberas, används oligo (dt) primers som specifikt kan binda till polyA-svansar för omvänd transkription, medan lncRNA och circRNA använder slumpmässiga hexamer (Random 6 mer) primers för omvänd transkription av totalt RNA. För miRNA används miRNA-specifika halsslinga omvända transkriptionsprimrar.Många företag har nu lanserat speciella tailing-kit.För stam-loop-metoden är tailing-metoden bekvämare, hög genomströmning och reagensbesparande, men effekten av att särskilja miRNA från samma familj bör inte vara lika bra som stam-loop-metoden.Varje omvänd transkriptionskit har krav på koncentrationen av genspecifika primrar (stam-loops).Den interna referensen som används för miRNA är U6.I processen med stam-loop-inversion, bör ett rör av U6 vändas separat, och de främre och bakre primers av U6 bör läggas till direkt.Både circRNA och lncRNA kan använda HKGs som intern referens.IcDNA-detektion,
om det inte är några problem med RNA bör cDNA också vara bra.Men om experimentets perfektion eftersträvas är det bäst att använda en intern referensgen (referensgen, RG) som kan skilja gDNA från cd-skivor.Generellt är RG en hushållsgen.HKG) såsom visas i figur 10;Vid den tiden gjorde jag lagringsprotein för sojabönor och använde actin7 innehållande introner som en intern referens.Storleken på det amplifierade fragmentet av denna primer i gDNA var 452 bp, och om cDNA användes som mall var det 142 bp.Sedan visade testresultaten att en del av cDNA:t faktiskt var kontaminerat av gDNA, och det visade också att det inte fanns några problem med resultatet av omvänd transkription, och det kunde användas som en mall för PCR.Det är värdelöst att köra agarosgelelektrofores direkt med cDNA, och det är ett diffust band, vilket inte är övertygande.
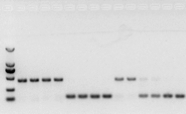
Fig 10 cDNA-detektion
Bestämning av qPCR-förhållandenär i allmänhet inga problem enligt protokollet för kitet, främst i steget tm-värde.Om vissa primrar inte är väl utformade under primerdesign, vilket resulterar i en stor skillnad mellan tm-värdet och den teoretiska 60°C, rekommenderas att cDNA Efter att proverna har blandats, kör en gradient-PCR med primers, och försök undvika att ställa in temperaturen utan band som TM-värdet.
Dataanalys
Den konventionella relativa fluorescens kvantitativa PCR-bearbetningsmetoden är i grunden enligt 2-ΔΔCT.Databehandlingsmall.
Relaterade produkter:
Realtids PCR enkeltTM –SYBR GREEN I
RT Easy I (Master Premix för första sträng cDNA-syntes)
RT Easy II (Master Premix för första sträng cDNA-syntes för qPCR)
Posttid: Mar-14-2023



