



Som ny i laboratoriet är det inget bra jobb att sålla bort positiva växter från ett gäng växter med låg konverteringsgrad.För det första måste DNA extraheras från ett stort antal prover en efter en, och sedan kommer de främmande generna att detekteras med PCR.Resultaten är dock ofta tomma och band med några få föremål ibland, men det är omöjligt att avgöra om det finns missade upptäckter eller falska upptäckter..Är det mycket hjälplöst att möta sådana experimentella processer och resultat?Oroa dig inte, bror lär dig hur du enkelt och exakt sållar bort transgena positiva växter.
Steg 1
Designdetekteringsprimers

Bestäm den endogena genen och den exogena genen som ska detekteras enligt provet som ska testas, och välj en representativ 100-500bp sekvens i genen för primerdesign.Bra primers kan säkerställa noggrannheten av detektionsresultaten och förkorta detektionstiden (se bilagan för vanliga detektionsprimers).
Observera: De nydesignade primrarna måste optimera reaktionsförhållandena och verifiera noggrannheten, precisionen och detektionsgränsen för detektionen innan storskalig detektering.
Steg 2
Designa experimentellt protokoll

Positiv kontroll: Använd det renade DNA som innehåller målfragmentet som mall för att avgöra om PCR-reaktionssystemet och -förhållandena är normala.
Negativ/tom kontroll: Använd DNA-mallen eller ddH2O som inte innehåller målfragmentet som mall för att detektera om det finns en kontamineringskälla i PCR-systemet.
Intern referenskontroll: använd primer/probkombinationen av den endogena genen i provet som ska testas för att utvärdera om mallen kan detekteras med PCR.
Lägga märke till:
Positiva, negativa/blanka kontroller och internkontrollkontroller bör ställas in för varje test för att utvärdera giltigheten av experimentresultaten.
Experimentförberedelse

Före användning, observera om lösningen är jämnt blandad.Om utfällning hittas måste den lösas och blandas enligt instruktionerna före användning.2×PCR-blandning måste pipetteras och blandas upprepade gånger med en mikropipett före användning för att undvika ojämn jonfördelning.
Lägga märke till:
Ta fram manualen och läs den noggrant, och gör förberedelser innan experimentet i strikt enlighet med manualens krav.
Steg 4
Förbered PCR-reaktionssystem

Enligt experimentprotokollet, blanda primrarna, H2O och 2×PCR-blandningen jämnt, centrifugera och fördela dem till varje reaktionsrör.
Lägga märke till:
För storskalig eller långtidstestning rekommenderas att använda ett PCR-reaktionssystem som innehåller UNG-enzym, vilket effektivt kan undvika aerosolkontamination orsakad av PCR-produkter.
Steg 5
Lägg till reaktionsmall

Med hjälp av Direct PCR-teknologi finns det inget behov av tråkig nukleinsyrareningsprocess, provmallen kan framställas inom 10 minuter och motsvarande PCR-reaktionssystem kan läggas till.
Lägga märke till:
Klyvningsmetoden har bättre detektionseffekt och den erhållna produkten kan användas för flera detektionsreaktioner.

5.1: Direkt expansion av löv
Beroende på storleken på bilden i manualen, skär bladvävnaden med en diameter på 2-3 mm och placera den i PCR-reaktionssystemet.
Obs: Se till att bladfragmenten är helt nedsänkta i PCR-reaktionslösningen och tillsätt inte överdriven bladvävnad.
5.2: Bladklyvningsmetod
Skär bladvävnaden med en diameter på 5-7 mm och placera den i ett centrifugrör.Om du väljer mogna löv, undvik att använda vävnaderna i bladets huvudven.Pipettera 50 ul buffert P1-lysat i ett centrifugrör för att säkerställa att lysatet helt kan sänka ner bladvävnaden, placera det i en termisk cykler eller ett metallbad och lysera vid 95°C i 5-10 minuter.

Tillsätt 50 ul buffert P2 neutraliseringslösning och blanda väl.Det resulterande lysatet kan användas som en mall och läggas till PCR-reaktionssystemet.
Obs: Mängden mall är mellan 5-10% av PCR-systemet och bör inte överstiga 20% (till exempel, i ett 20μl PCR-system, tillsätt 1-2μl lyslösning, inte mer än 4μl).
Steg 6
PCR-reaktion

Efter centrifugering av PCR-reaktionsröret placeras det i ett PCR-instrument för amplifiering.
Lägga märke till:
Reaktionen använder icke-renad mall för amplifiering, så antalet amplifieringscykler är 5-10 fler cykler än när man använder renad DNA-mall.
Steg 7
Elektroforesdetektering och resultatanalys
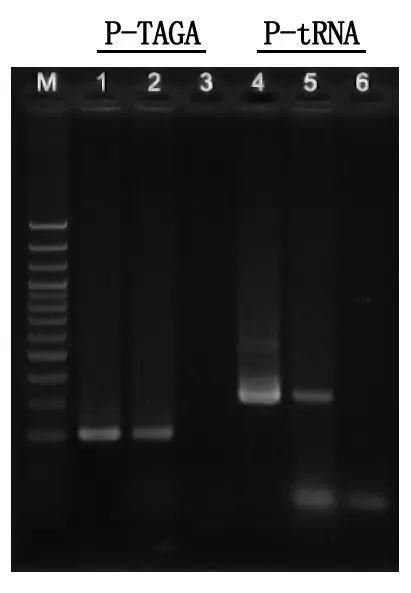
M: 100 bp DNA-stege
1\4: Renad DNA-metod
2\5: Direkt PCR-metod
3\6: Tom kontroll
QC:
Testresultaten för de olika kontrollerna i experimentet bör uppfylla följande villkor.Annars bör orsaken till problemet analyseras, och testet bör utföras igen efter att problemet har åtgärdats.
Tabell 1. Normala testresultat för olika kontrollgrupper
*När plasmiden används som en positiv kontroll kan det endogena gentestresultatet vara negativt
Resultatbedömning:
A. Testresultatet för den endogena genen i provet är negativt, vilket indikerar att DNA som är lämpligt för vanlig PCR-detektion inte kan extraheras från provet eller så innehåller det extraherade DNA:t PCR-reaktionshämmare, och DNA:t bör extraheras igen.
B. Testresultatet för den endogena genen i provet är positivt, och testresultatet för den exogena genen är negativt, vilket indikerar att DNA som är lämpligt för vanlig PCR-detektion extraheras från provet, och det kan bedömas att XXX-genen inte detekteras i provet.
C. Testresultatet för den endogena genen i provet är positivt och testresultatet för den exogena genen är positivt, vilket indikerar att DNA som är lämpligt för vanlig PCR-detektion har extraherats från provet och att provets DNA innehåller XXX-genen.Bekräftelseförsök kan utföras ytterligare.
Steg 8
Designdetekteringsprimers

Efter experimentet, använd 2% natriumhypokloritlösning och 70% etanollösning för att torka av experimentområdet för att förhindra miljöföroreningar.
Posttid: 2021-08-08



